Theorie

Das Helmholtz-Institut Münster bildet gemeinsam mit dem Institut für Physikalische Chemie der Westfälischen Wilhelms-Universität Münster ein interdisziplinäres Team für theoretische Untersuchungen im Bereich der Elektrolytforschung.
Methoden
Die Methoden der Gruppe für Theorie umfassen klassische Molekulardynamik-Simulationen (MD), quantenchemische Berechnungen (hauptsächlich DFT), ab initio MD-Simulationen sowie Monte-Carlo-(MC)-Ansätze. MD-Simulationen werden verwendet, um die Struktur und den Transport in großen Elektrolytsystemen mit atomistischer Auflösung zu untersuchen. Quantenchemische Ansätze ermöglichen die Charakterisierung der elektrochemischen Eigenschaften einzelner molekularer Komponenten. Mit ab initio MD-Berechnungen können zum Beispiel Reaktionsgeschwindigkeiten abgeschätzt werden. MC-Simulationen werden unter anderem für eine grobkörnige Darstellung von Batterien verwendet.
Ein kürzlich entwickeltes MD/MC-Hybridschema ermöglicht die Behandlung von chemischen Reaktionen in klassischen MD-Simulationen. Zusätzlich werden Konzepte des maschinellen Lernens entwickelt und zur Erzeugung von quantenmechanisch (DFT) basierten Kraftfeldern angewendet.
Harmonische Systeme für mehr Funktionalität
Die vom Team beforschten Systeme umfassen Polymer- und Flüssigelektrolyte einschließlich (multifunktionaler) Additive und komplexere Hybridsysteme, die so abgestimmt sind, dass sie fortgeschrittene Elektrolytfunktionalitäten ermöglichen. Die Systeme werden entweder im Volumen oder in der Nähe von Elektrodengrenzflächen untersucht. Darüber hinaus werden Anoden, wie metallisches Lithium mit möglicher Dendritenbildung, und polymere Kathodensysteme erforscht.
Umfassende Charakterisierung
Mehrere konzeptionelle Entwicklungen der Theorie-Gruppe sind für ein besseres Verständnis der mikroskopischen Mechanismen von großer Bedeutung:
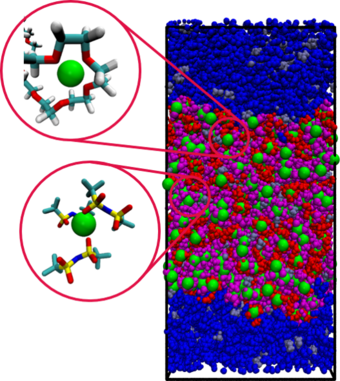
(1) Für den Ionentransport in polymeren Elektrolyten wurde ein Transportmodell entwickelt, das auf der Polymerphysik basiert und die verschiedenen Mechanismen der Kationendynamik erfasst.
(2) Der Einfluss von Impulserhaltung und hydrodynamischer Wechselwirkung auf den Ionentransport und insbesondere die Konsequenzen für die resultierende Leitfähigkeit werden untersucht. Dies beinhaltet auch neue Ansätze zur Beschreibung der Relevanz des Transports über vehikuläre vs. Sprungmechanismen.
(3) In bestimmten Situationen können starke elektrische Felder zu nichtlinearem Ionentransport führen.
Durch ihre geeignete Charakterisierung können neue Informationen über Transporteigenschaften gewonnen werden.
Interdisziplinäre Kollaborationen
Die Kollaborationen des Theorie-Teams umfassen die Interaktion mit experimentellen Gruppen, was zu einem besseren Verständnis der experimentellen Ergebnisse führt. Gleichermaßen trägt sie zu einer Verbesserung der theoretischen Basis bei. Andererseits erlaubt der Austausch mit Modellierungsgruppen von der eher makroskopischen Seite die Etablierung echter Multiskalenansätze zum Verständnis der detaillierten Mechanismen. Ein Beispiel stellt die Batterie als Ganze dar, die hauptsächlich auf rein mikroskopischen Informationen beruht.
Publikationen:
Physical Chemistry Chemical Physics 2020, 22, 525, DOI: 10.1039/C9CP04947A
Applied Materials & Interfaces 2020, 12, 567, DOI: 10.1021/acsami.9b16348
ChemElectroChem 2020, 7(6), 1499-1508, DOI: 10.1002/celc.202000386
Chemistry of Materials 2019, 31(9), 3118-3133, DOI: 10.1021/acs.chemmater.8b04172
Journal of Physical Chemistry C 2018, 122(38), 21770-21783, DOI: 10.1021/acs.jpcc.8b06560
Batteries 2018, 4(4), 62, DOI: 10.3390/batteries4040062
Weitere Informationen finden Sie hier:
Ansprechpartner
