Prionkrankheiten
Prionkrankheiten oder transmissible spongiforme Enzephalopathien (TSE) sind eine spezielle Gruppe von neurodegenerativen Erkrankungen. Sie treten beim Menschen - Creutzfeldt-Jakob-Krankheit (CJD für "Creutzfeldt-Jabob disease") sowie bei Tieren - u.a. "Scrapie" bei Schaf, "bovine spongiform encepahlophathy" (BSE) bei Rindern auf. Prionkrankheiten können einen spontanen, genetischen oder infektiösen Hintergrund haben. Die Übertragbarkeit durch Infektion unterscheidet Prionkrankheiten von anderen neurodegenerativen, wie z.B. die Alzheimersche Demenz oder Chorea Huntington. Im Krankheitsverlauf treten Verhaltensänderungen, koordinative Fehlfunktionen (Ataxie) und Demenz auf neuropathologisch sind schwammartige (spongiforme) Änderungen im Gehirn, Degeneration von Neuronen und Astrocytose als histopathologische Kennzeichen von Prionkrankheiten nachweisbar (Abb. 1).
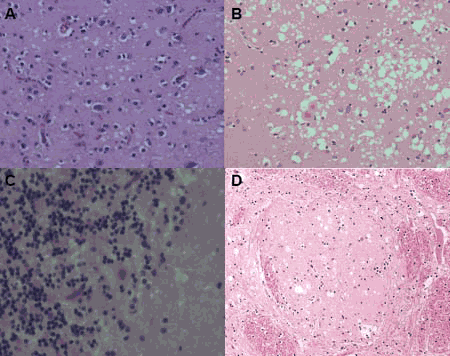
1982 veröffentlichte Stanley B. Pruisiner die Prion-Hypothese, welche besagt, dass der Erreger der Prionkrankheiten ausschließlich aus proteinösen Partikeln (Prionen) bestand. Der Hauptbestandteil der Prionen ist das Prion Protein in einer fehlgefalteten Struktur (PrPSc). Das Prion Protein ist ein körpereigenes Protein (PrPC). Der Replikationsmechanismus beruht darauf, dass PrPSc in der Lage ist mit PrPC zu interagieren und PrPC in PrPSc zu überführen.
Wir entwickeln eine Methode zu frühen Diagnose von Prionkrankheiten, indem wir den Aggregationszustand als Biomarker nutzen.
Zusätzlich untersuchen wir den zugrunde liegenden Replikationsmechanismus in vitro.